科学普及

从药物靶点发现到一期临床试验
1、 创新药物研发的流程
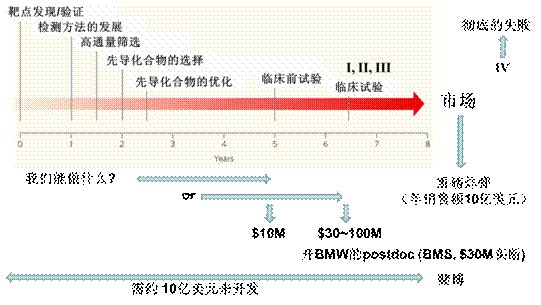
图一: 创新药物研发的流程
新药的研发,涉及多种学科和领域。近年来,随着分子生物学,药物化学,分子药理学,计算化学等等学科的发展,新药的研发已经从最初的以经验性为主向理论指导下进行药物分子设计方向发展。其开发流程为:首先选定药物作用的靶点,生物靶点的选定是研制药物的起步点。分子生物学上发现的一些新颖的重要的酶和受体等都将成为研制独特作用机理的药物的新靶点。靶点选定后,需要建立对其作用可评价的检验测定的生物模型,一般开始是用体外方法,在分子水平、细胞水平,或离体器官进行活性评价,在此基础上用实验动物的病理模型进行体内试验。检验方法确定后,我们就可以进行高通量筛选,从而确定先导化合物。该先导化合物可能特异性不高,药代动力学性质不合理或毒性较大,不能直接药用。因此我们可以将其作为新的结构类型和线索物质,并通过计算机辅助进行优化设计,对其结构进行修饰和改造,即先导化合物的优化,以使生物学性质臻于完善,达到安全、有效和可控的药用目的。从这些优化的化合物中选出候选物进行临床前研究,再进行I、II、III期临床研究。近年来,新药的研究与开发虽然更理性,但是成功率仍很低。创制一个全新药物并上市,一般需要从进行临床三期研究的2.5个候选物中得到,后者则需要6.5个化合物进行一期临床研究,为此,要在21个进行了慢性毒性的化合物中选取,这又需要合成6200个化合物,这全过程需时13年,耗资约13亿。
2、药物候选物AT-406的研发过程
|
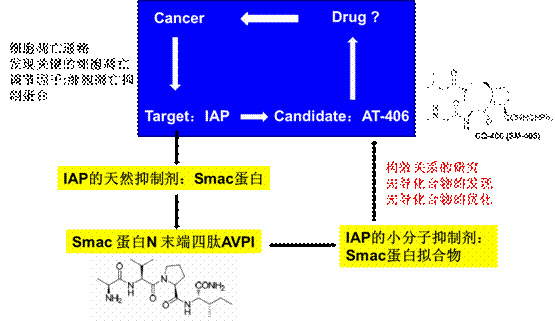

图二:AT-406的研发过程
药物候选物AT-406的研发过程也大致是上述一个过程。首先细胞凋亡通路的阐明,发现关键的细胞凋亡调节因子:细胞凋亡抑制蛋白。这类蛋白均具有高度保留的BIR(Baculovirus IAP Repeat)活性区域,直接用于抑制Caspase 的活性。肿瘤细胞通过过度表达IAP 从而抑制肿瘤细胞的正常调亡,导致肿瘤的疯狂增长。在目前所发现的八个细胞凋亡抑制蛋白中,XIAP(X-chromosome linked IAP)、c-IAP1(cellular IAP 1)和c-IAP2(cellular IAP 2)是细胞凋亡抑制活性最强的三个成员。如图1 所示,c-IAP1/2 通过抑制caspase-8/10 的激活,从而抑制细胞凋亡的外部通路的顺利进行;另一方面,XIAP 通过抑
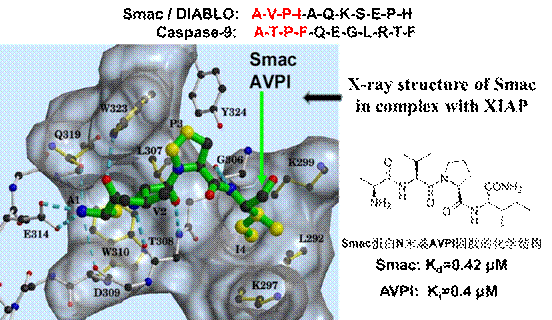
图三 Smac蛋白N末端AVPI四肽与XIAP结合的X-ray结构图
制caspase-9 和caspase-3/7,既可以抑制细胞凋亡的外部通路,亦可以阻断抑制细胞凋亡的内部通路。而由线粒体所释放的Smac蛋白是IAP的天然抑制剂。成熟的Smac 蛋白和XIAP 的BIR3 活性区域的结合常数Kd与其N 末端四肽(AVPI)和XIAP 的BIR3 活性区域的结合常数均为400 nM;说明我们可以用小分子模拟AVPI 四肽,即小分子
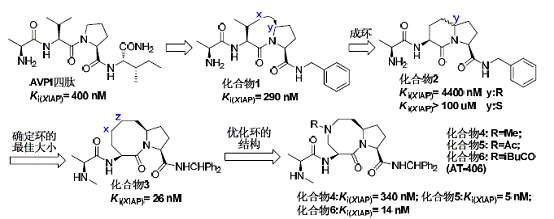
图四 单体小分子Smac 拟合物的发展
Smac 蛋白拟合物,来代替整个成熟的Smac 蛋白与XIAP 作用,起到促进肿瘤细胞凋亡的作用。因此,我们以Smac 的N 末端四肽作为小分子Smac 蛋白拟合物的设计起始模板,首先设计合成了图四的化合物1,在经过成环、确定环的大小、优化环的结构等一系列过程,最终在合成的几百个化合物寻找到活性最佳、口服生物利用度高、毒性低的AT-406作为药物候选物。现在AT-406已经进入临床I期研究,并且在2011年9月7日,瑞士制药公司以1000万美元买断AT-406的相关专利和后续发展权。但是我们可以看到,AT-406距离最终上市的道路还很漫长,我们期待其后续的发展。
附件下载:
